PREP MATERIAL
Stage 19
The structure of clofarabine (2-chloro-2'-fluro-deoxy-9-β-D-arabinofuranosyladenine) incorporates some of the favorable anti-tumor properties of fludarabine and cladribine.1 The chloride chain in the adenosine ring provides resistance to deamination by adenosine deaminase; a fluoride in the 2' position of the arabinofuranosyl ring hinders phosphorolysis by purine nucleoside phosphorylase (PNP) (Figures 5A and 5B).
Figure 5A. Clofarabine - Resistant to Deamination & Phosphorolysis.
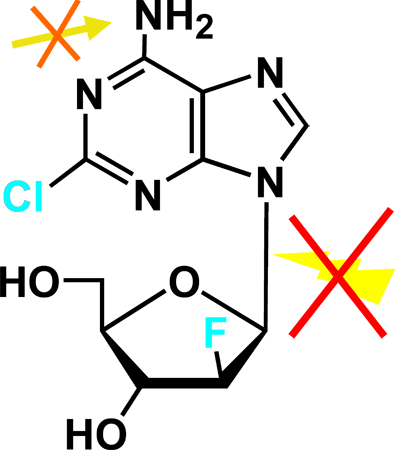
Figure 5B. Deoxyadenosine Analogs.
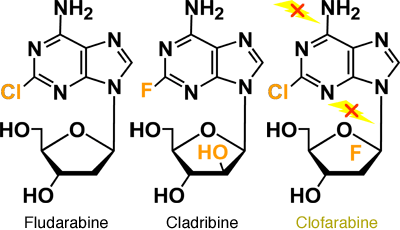
Nucleoside analogues (purine:adenosine, guanosine/pyrimidine:thymidine/cytosine) are one of the most widely used classes of drugs for the treatment of cancer, and have been a rich source of active agents in hematologic malignancies, especially the leukemias. A number of new nucleoside analogs are emerging with additional metabolic properties and mechanisms of action and their activity is currently being evaluated in clinical trials. One of these nucleosides is clofarabine, a second-generation deoxynucleoside analog which was designed as a rational extension of older analogs such as fludarabine or cladribine.
The molecular structure of clofarabine is mainly determined by the addition of two halogen groups. These two halogen groups are responsible for the better stability compared to other nucleosides. In addition, clofarabine is a good example of how supposedly minor changes of the molecular structure can lead to significantly different mechanisms of action and also a different spectrum of activity.
Clofarabine is phosphorylated by dCK to its active metabolite, clofarabine triphosphate, resulting in the inhibition of DNA polymerases and ribonucleotide reductase (RR), enzymes important in nucleoside analog metabolism. Clofarabine is currently under investigation in AML as a single agent or in combination with other chemotherapeutic agents.
Inhibition of Ras signal transduction is of interest in AML and MDS.2 Ras-mediated signaling can be inhibited by prevention of its membrane localization. The C-terminal prenylation of Ras, required for its association with the cell membrane and its transforming activity, is mediated by the enzyme farnesyl transferase (Ftase) (Figure 6).
Figure 6. Farnesyl Transferase Inhibitors.
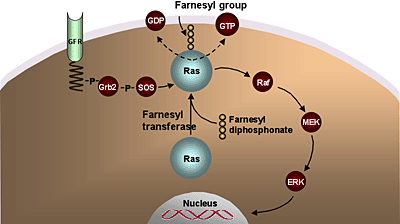
Inhibition of Ras signal transduction remains of interest in AML and MDS, as mutations and dysregulations of Ras have been associated with the development of myeloid leukemias. Ras-mediated signaling can be inhibited by prevention of its membrane localization, inhibition of Ras protein expression using antisense nucleotides or inhibition of its downstream targets.
Several pharmacologic inhibitors of FTase have been developed and are in clinical trials. It is important to realize, though, that these agents may have effects on other signaling components independent of Ras, as responses do not correlate with the mutations status of Ras.
Several pharmacological inhibitors of Ftase, such as tipifarnib, have been developed and some have undergone investigation in clinical trials.3 In a study of 158 elderly patients treated with tipifarnib, 14% of patients experienced a CR with a median duration of 7.3 months and median survival of 18 months. The ability to effectively target AML in a relatively resistant population was encouraging.
CD33 is expressed on the surface of more than 90% of AML cells with an average antigen density of 10,000 sites per cell. CD33 is expressed on normal granulocyte/monocyte colony forming units (CFU-GM) and some primitive erythroid progenitors. However, its expression on tissues other than the hematopoietic system and the normal pluripotent hematopoietic stem cells is not prominent. Gemtuzumab ozogamicin, a conjugate of a CD33 antibody to calicheamicin, has been approved by the FDA for the treatment of elderly patients (>60 years) with AML in first relapse and first CR duration >3-6 months.4 A multicenter study of 142 patients with AML in first relapse treated with gemtuzumab ozogamicin 9 mg/m2 on days 1 and 15 resulted in a CR rate of 16% and an overall response rate of 30%, including patients without evidence of leukemia but with incomplete hematopoietic recovery. Its use is also being explored as a part of combination chemotherapy with standard cytotoxic agents.
Suppression of programmed cell death (apoptosis) has been demonstrated in myeloid as well as lymphoid malignancies.5 Dysregulation of apoptosis prolongs leukemic cell survival, resulting in their expansion independently of cell division. Creation of a permissive environment for genetic instability and accumulation of gene mutations, contribute further to the process of neoplastic change. Defects of apoptotic machinery promote resistance to immune-based destruction, facilitate growth factor-independent cell survival and confer resistance to cytotoxic agents. Such resistance is an important factor determining the likelihood of response. Thus, down-regulation of anti-apoptotic proteins such as Bcl-2 may reduce the threshold for chemotherapy resistance and restore chemosensitivity to leukemia cells. Genasense is an 18-mer phosphorothioate oligodeoxynucleotide antisense designed to bind the first 6 codons of human Bcl-2 mRNA.6 A recent study combining genasense with chemotherapy to treat untreated patients older than 60 years did not demonstrate a superior outcome to chemotherapy alone.
A number of oncogenes with constitutive kinase activity have been described as attractive therapeutic targets. Mutations that remove inhibitory domains of the molecule or induce the kinase domain to adopt an activated configuration lead to the constitutive activation of the protein product. Aberrations of genes including c-ABL, c-FMS, FLT3, c-KIT, PDGFRα and PDGFRβ, normally involved in the regulation of hematopoiesis, have been described.7 As a result, a number of downstream signaling cascades such as the Jak-Stat pathway, the Ras/Raf/MAPK pathway and the PI3K pathway are activated leading to inappropriate cell proliferation and survival (Figure 7).
Figure 7. JAK-STAT Signaling Pathway.
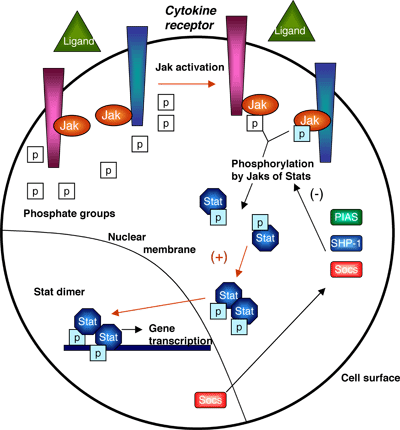
Despite the pivotal role of these pathways in normal cellular function, their inhibition may not be associated with significant clinical toxicity, as they are inappropriately activated in leukemic cells. Therefore, their partial inhibition may be sufficient to interfere with malignant cell growth. A number of kinase inhibitors have been investigated for treating patients with myeloid neoplasms.
References
- Kantarjian HM, Gandhi V, Kozuch P, et al. Phase I clinical and pharmacology study of clofarabine in patients with solid and hematologic cancers. J Clin Oncol. 2003;21:1167-1173.
- Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062-1074.
- Lancet JE, Gojo I, Gotlib J, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109:1387-1394.
- Sievers EL, Larson RA, Stadtmauer EA, et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J Clin Oncol. 2001;19:3244-3254.
- Reed JC. Dysregulation of apoptosis in cancer. J Clin Oncol. 1999;17:2941-2953.
- Marcucci G, Byrd JC, Dai G, et al. Phase 1 and pharmacodynamic studies of G3139, a Bcl-2 antisense oligonucleotide, in combination with chemotherapy in refractory or relapsed acute leukemia. Blood. 2003;101:425-432.
- Scheijen B, Griffin JD. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene. 2002;21:3314-3333.